Immunoprecipitation (optimization?) problem - anti-ovalbumin lane has 225 kDa band. (Feb/03/2009 )
Hi,
I am learning how to do IPs, and am having a lot of trouble. Could someone please help me troubleshoot my procedure? I am optimizing a protocol to IP PBMC surface markers using our own mAbs, and need some expert advice. I have included photographs of 2 coomassie-stained gels.
I started with our own mAbs, but got inconclusive results. (We know the mw of the proteins two of them pull down, and want the mw of the other three.) To test my protocol, I then tried spiking a PBMC cell lysate with .01 ug/ml ovalbumin and a commercially-available anti-oval antibody; when that worked, I then used a cell lysate and tried pulling down CD45 using one of our hybridoma SNs. I get a band in the CD45 lane, but also a matching band in the CD45 lane.
Our mAbs and commercial anti-oval:

Our anti-CD45 and commercial anti-oval:
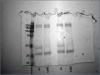
I don't think it's overflow. I am not getting much guidance from my prof, and he says that there are no biochemists at our uni that can help me. Could someone on the BB help me, please?
Thank you.
While I'm not exactly sure what you are doing and what your problem is, I know that
many problems (not the one I posted above!) can be solved by doing 2 things.
#1. Do a negative control IP with an IgG. This will indicate background. If you have background optimize washing and preclearing.
#2. If you IP with a mouse antibody and Western with a mouse antibody you will get artifacts! Use a different isotype for each step.
PS. the bands you are seeing around 45 kDA are IgGs (there is a heavy IgG that runs around 50 and a light that runs around 35kDa). Do a negative control with a mouse IgG and this will shed much light on the situation.
Hi Mike,
Thanks for your reply. Sorry, I got interrupted while posting this and didn't get my protocol up here.
Here is my protocol:
1. Lysis of PBMC using 300 ul non-denaturing lysis buffer.
(20 mM Tris-HCl, pH 8, 137 mM NaCl, 10% glycerol, 1% TX-100, 2 mM EDTA, protease inhibitors)
2. Mix together mAB and DPBS+protease inhibitors.
(to get 50 ug of mAb)
3. Add mAb to washed Protein G beads (bead=50 ul of 50% slurry). Incubate until PBMC lysis is complete.
4. After lysis is complete, wash excess mAb off beads with DPBS+protease inhibitors.
5. Add 200 ul DPBS+1% NP-40+protease inhibitors to 300 ul lysate.
5. Add 250 ul lysate to each tube of beads.
6. Incubate at RT for 2 hr with shaking.
7. Wash beads 2X with non-denaturing lysis buffer. Save washes for analysis.
8. Add 50 ul Sample buffer to beads.
9. Boil beads. Place on ice to cool. Spin to pellet beads. Load SN on gel.
I haven't tried WB yet. I have been staining with Coomassie to visualize the proteins on the gel to make sure that my procedure works. Could my protein load be too small to visualize with staining? I figured that my 50 & 25 kDa bands were my light and heavy chains, but the 200 kDa protein in the antiovalbumin well baffles me. Ovalbumin runs about 45 kDa, as well.
Oops, I meant to say a matching band in the antiovalbumin lane.
Both anti-ovalbumin and anti-CD45 seem to pull down a protein with the same molecular weight. This is the part I am confused about...
Hi again,
The protein you IP will not necessarily be observable by Coomassie blue. In fact, it is rarely observable unless you IP 2mg proteins or more and have a good antibody. You need to do a Western to determine how good your Ips are.
You will always IP many proteins. An IP will pull down all the interacting proteins associated with your protein during an IP. The Coomassie will not tell you much information (until your IPs are highly efficient and optimized).
mikew on Feb 6 2009, 05:40 PM said:
The protein you IP will not necessarily be observable by Coomassie blue. In fact, it is rarely observable unless you IP 2mg proteins or more and have a good antibody. You need to do a Western to determine how good your Ips are.
You will always IP many proteins. An IP will pull down all the interacting proteins associated with your protein during an IP. The Coomassie will not tell you much information (until your IPs are highly efficient and optimized).
Hi Mike,
Got it. Your answer helps a lot! Thanks.
S.
Update:
After struggling so much with the IPs, I changed tack. I decided to try BN-PAGE instead, using Schagger's method as a guide.
So far, so good...I have tried labeling whole cell lysate BN-PAGE blots with 5 of our antibodies, and three of them seem to work OK. I'm looking forward to getting the other 10 antibodies to work!