Gel Purification problem..Help~! - (Jul/06/2009 )
Dear ev1~
Need help to figure out my gel purification problem..
Here's my long long story..
(1) I loaded 40microlit of PCR product sample on the gel and managed to get a nice intense band on the gel, then i cut the band out and purified it using Qiaquick purification kit. But after purified, i run the purified product on the gel electrophoresis to check the quality..only a very faint band can be seen..compared to my original band before cutting. Can anyone explain what went wrong? I followed the protocol exactly.
(2) Some suggested i re-run PCR using the purified PCR product as template. I tried and good news i got even thicker expected band but bad news is it also has other faint bands formed with primer dimer. Some suggested that it maybe cause cutting gel technique when cutting gel, may have accidentally cut the nearby band and suggested i shld run the gel electrophoresis for longer time to separate the gap of my expected band with other bands.
Anyone can recommend me or suggest what i can alter and optimize? What voltage or how long to run (* just dont want my gel to melt before i cut it or over run it~) Currently, i'm running at 90V for 50minutes.
Any suggestions are welcome..Thanks ^^
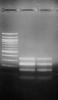
Here i include the scanned gel pics as reference. (1) bfore purified (2) after purified at position 250bp![]()
7iew on Jul 6 2009, 03:58 PM said:
Need help to figure out my gel purification problem..
Here's my long long story..
(1) I loaded 40microlit of PCR product sample on the gel and managed to get a nice intense band on the gel, then i cut the band out and purified it using Qiaquick purification kit. But after purified, i run the purified product on the gel electrophoresis to check the quality..only a very faint band can be seen..compared to my original band before cutting. Can anyone explain what went wrong? I followed the protocol exactly.
(2) Some suggested i re-run PCR using the purified PCR product as template. I tried and good news i got even thicker expected band but bad news is it also has other faint bands formed with primer dimer. Some suggested that it maybe cause cutting gel technique when cutting gel, may have accidentally cut the nearby band and suggested i shld run the gel electrophoresis for longer time to separate the gap of my expected band with other bands.
Anyone can recommend me or suggest what i can alter and optimize? What voltage or how long to run (* just dont want my gel to melt before i cut it or over run it~) Currently, i'm running at 90V for 50minutes.
Any suggestions are welcome..Thanks ^^
Here i include the scanned gel pics as reference. (1) bfore purified (2) after purified at position 250bp
1) How much of your purified product do you load in the second gel to confim? QiaGen kits are known for they inefficiency, you#ll never recover all your product. Which volume do you use to elute your product? You can improve recovery by warming up the buffer and also reducing the volume to ~30µl.
2) I'd definitely run the gel for a bit longer to separate your specific band from the unwanted ones.
Finally, what do you want the PCR product for? Depending on the amount you've loaded on the gel, and your total amount, you might actually have enough!!
7iew on Jul 6 2009, 09:58 AM said:
Need help to figure out my gel purification problem..
Here's my long long story..
(1) I loaded 40microlit of PCR product sample on the gel and managed to get a nice intense band on the gel, then i cut the band out and purified it using Qiaquick purification kit. But after purified, i run the purified product on the gel electrophoresis to check the quality..only a very faint band can be seen..compared to my original band before cutting. Can anyone explain what went wrong? I followed the protocol exactly.
(2) Some suggested i re-run PCR using the purified PCR product as template. I tried and good news i got even thicker expected band but bad news is it also has other faint bands formed with primer dimer. Some suggested that it maybe cause cutting gel technique when cutting gel, may have accidentally cut the nearby band and suggested i shld run the gel electrophoresis for longer time to separate the gap of my expected band with other bands.
Anyone can recommend me or suggest what i can alter and optimize? What voltage or how long to run (* just dont want my gel to melt before i cut it or over run it~) Currently, i'm running at 90V for 50minutes.
Any suggestions are welcome..Thanks ^^
Here i include the scanned gel pics as reference. (1) bfore purified (2) after purified at position 250bp
For gel purifications using Qiagen, I consider it a good day if I get 50% yield. Even the PCR or Enzyme cleanup kits usually top out around 70%, at least for me.
You could run your gel for longer, but at a slower speed to avoid running it too hot to get better separation. However, it may just be an inherent problem with the purification kit. If you need a more concentrated purification, you could try the Minelute kit rather than the Qiaquick kit. You can elute in as little as 10 ul with the minelute (and your product must be < 4 kb).
But considering your PCR product is 250 bp, my guess is that while you may have lost a little in the concentration from the beginning, you probably still have enough copies of your product to do what you need to do... it just depends on what you need to do.
Do you know what the concentration of the purified product is?
My first question would be "Why do I have so many bands on my PCR?". Once I'd solved that problem -- perhaps by re-designing the primers -- I'd worry about the gel purification problems.
With well-designed primers you'd probably get three or four times greater quantity of the specific band you're after, and your gel purification problems will likely dissappear.
Failing that, I agree with the others -- you've probably got enough DNA to proceed, depending on what it is you want to do with it.
7iew on Jul 6 2009, 10:58 PM said:
Need help to figure out my gel purification problem..
Here's my long long story..
(1) I loaded 40microlit of PCR product sample on the gel and managed to get a nice intense band on the gel, then i cut the band out and purified it using Qiaquick purification kit. But after purified, i run the purified product on the gel electrophoresis to check the quality..only a very faint band can be seen..compared to my original band before cutting. Can anyone explain what went wrong? I followed the protocol exactly.
(2) Some suggested i re-run PCR using the purified PCR product as template. I tried and good news i got even thicker expected band but bad news is it also has other faint bands formed with primer dimer. Some suggested that it maybe cause cutting gel technique when cutting gel, may have accidentally cut the nearby band and suggested i shld run the gel electrophoresis for longer time to separate the gap of my expected band with other bands.
Anyone can recommend me or suggest what i can alter and optimize? What voltage or how long to run (* just dont want my gel to melt before i cut it or over run it~) Currently, i'm running at 90V for 50minutes.
Any suggestions are welcome..Thanks ^^
Here i include the scanned gel pics as reference. (1) bfore purified (2) after purified at position 250bp
1) It is pretty normal for you to see a fainter band as compared to your original band. I have had cases whereby my band was so faint after purification that it looked as though it wasn't there, but still, it was enough to go on for other things. We normally do not get back everything that we purify as the column has its limits. To increase binding of the DNA to your column, increase the amount of the binding buffer, ie, QG in the first step or the optional step. You can also warm up your water to 50degcel before you use it for elution, it helps in the yield. Alternatively, what you can do is to take the eluate after the elution step, and deposit it in the column again, let it stand for an additional minute, den elute again.
2) It is pretty normal at times that you get other bands for your PCR after gel purification. You definitely should run the gel electrophoresis for a longer time, but at a lower voltage, say... 80V for 2 hours or 3 hours, till the band you want is 1 or 1.5 inch above the end of the gel. Cut it out and purify it and do a PCR for it.
Hi almost a doctor~,
1) I loaded 5μl of sample + 1 loading dye into the well to confirm. I tried 30μl but still faint.
2) I tried run for 1hour to separate them well. it works~
Finally, i nd my PCR product to send for sequencing. If the band is not visible seen, probably i wont able to seq my expected band out.
HomeBrew on Jul 7 2009, 05:48 AM said:
With well-designed primers you'd probably get three or four times greater quantity of the specific band you're after, and your gel purification problems will likely dissappear.
Failing that, I agree with the others -- you've probably got enough DNA to proceed, depending on what it is you want to do with it.
Hi ' Home Brew',
I'm using degenerate primers to get my expected band. It is because the gene from this species of my sample is still not been researched on and based on previous researches, there is a range of the sizes for the expected band for the gene i nd to find which is done from a group of the same genus but differ. species.
Probably i'm still new in this work and many stuffs i still unsure regarding abt this primers set which previously designed by my senior. I'm doing a continuation work from her.
I nd to get those bands for sequencing to see which is the my expected gene. which falls within this range of band sizes.
Got anymore suggestions to help me thru? THanks~

fishdoc on Jul 7 2009, 12:04 AM said:
7iew on Jul 6 2009, 09:58 AM said:
Need help to figure out my gel purification problem..
Here's my long long story..
(1) I loaded 40microlit of PCR product sample on the gel and managed to get a nice intense band on the gel, then i cut the band out and purified it using Qiaquick purification kit. But after purified, i run the purified product on the gel electrophoresis to check the quality..only a very faint band can be seen..compared to my original band before cutting. Can anyone explain what went wrong? I followed the protocol exactly.
(2) Some suggested i re-run PCR using the purified PCR product as template. I tried and good news i got even thicker expected band but bad news is it also has other faint bands formed with primer dimer. Some suggested that it maybe cause cutting gel technique when cutting gel, may have accidentally cut the nearby band and suggested i shld run the gel electrophoresis for longer time to separate the gap of my expected band with other bands.
Anyone can recommend me or suggest what i can alter and optimize? What voltage or how long to run (* just dont want my gel to melt before i cut it or over run it~) Currently, i'm running at 90V for 50minutes.
Any suggestions are welcome..Thanks ^^
Here i include the scanned gel pics as reference. (1) bfore purified (2) after purified at position 250bp
For gel purifications using Qiagen, I consider it a good day if I get 50% yield. Even the PCR or Enzyme cleanup kits usually top out around 70%, at least for me.
You could run your gel for longer, but at a slower speed to avoid running it too hot to get better separation. However, it may just be an inherent problem with the purification kit. If you need a more concentrated purification, you could try the Minelute kit rather than the Qiaquick kit. You can elute in as little as 10 ul with the minelute (and your product must be < 4 kb).
But considering your PCR product is 250 bp, my guess is that while you may have lost a little in the concentration from the beginning, you probably still have enough copies of your product to do what you need to do... it just depends on what you need to do.
Do you know what the concentration of the purified product is?
Hi fishdoc~,
I need my expected band for sequencing. I need to know whether the expected band is my expected seq for the gene i wanted. Due to my expected gene size is ranging from 150-300bp. Thus, i nd to cut out those bands in those range to seq. to see which is my desired gene.
Hmm..i dun really know for my conc. of my purified prod. I have limited amt of purified prod sample so i didn run a spectro on it to check it conc. but by the looks of it in the gel comparing with ladder, my expected band wasnt tht clear though..
