Explanation needed for melting curve - (Mar/21/2012 )

Hi,
I'm very new to qPCR.
I'm using SYBR green to detect various genes (using total RNA), housekeeping (GAPDH) seems to give good results but the problem is the other few genes they keep giving me funny results, i.e. slope > -2.0 though I managed to get R^2 ~ 0.95. After some readings I figured out I should've done the melting curve first to detect primer-dimers formation, so I did.
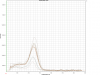
Attached example here is the melting curve for one gene. Due to the funny results after few attempts previously, I chucked the primer and use the new stock for this run. The samples mostly give Tm~82.5degC, but all standards have lower Tm ~72.5degC. I supposed the lower-Tm amplicons are primer-dimers, but how come the samples do amplify correctly, but none of the standards? What could be the reasons for this, and how do I improve my qPCR?
Thnx
-ctfazra-
Sounds like you need to make new standards using the end-product of your "good" qPCR run. Just Qiaquick purify what is in the well and make a dilution series in TE buffer with SSS dna.
do u mean if qPCR for GAPDH works well, then i need to get the plate and take out the remaining standards i have from the wells, purify and use them again for another gene?
No. I mean that it seems like your GAPDH "standards" are the amplification of something different than the GAPDH PCR product that you are successfully amplifying in your samples. You just need to make new standards. To do this, get the plate, take out the 20ul of finished qPCR reaction from one of your samples that gives a beautiful melting curve, and then purify the PCR product using a kit (e.g. Qiaquick). Then measure the DNA with a nanodrop, and dilute it 1:10, 1:100: 1:1000: 1:10000, etc. in TE buffer with 10ug/ml sheared salmon sperm DNA. Then these are your new standards. To be certain, you should send the PCR product generated from your old standards and the one generated using these new standards for sequencing.